Research Highlights
The central hypothesis of our laboratory is that the isoform-level gene products “transcript variants” and “protein isoforms” are the basic functional units in the mammalian cell, and accordingly, the informatics platforms – ranging from basic molecular biology data management systems to the biomarker and therapeutic drug target discovery for precision medicine – should adapt “gene-isoform-centric” rather than “gene-centric” approaches.
Our research is focused on developing statistical machine learning based algorithms and informatics solutions for knowledge extraction in biology and medicine. Our ongoing research work, funded by National Library of Medicine/NIH R01, focuses on developing novel bioinformatics methods to help in silico discovery and research for accelerating the linkage of phenotypic and genomic information at gene-isoform level. The overarching goal of the lab is to translate data from high dimensional (-omic) platforms (e.g., NextGen sequencing) to derive experimentally interpretable and testable discovery models for the identification and characterization of transcript-variants/protein isoforms, networks and pathways involved in normal and disease cells.
Viral DNABERT: A robust BERT model for SARS-Cov2 and other virus strains
Code will be available soon
Pratik Dutta, Zhihan Zhou
Our group used the existing DNABERT model and science and applied it to virus strains (in collaboration with Dr Han Liu, Department of Computer Science, Northwestern University).
DeepMOIS-MC: Deep Multi-Omics Integration by Learning Correlation-Maximizing for Cancer Subtyping
Code will be available soon
Yanrong Ji, Pratik Dutta
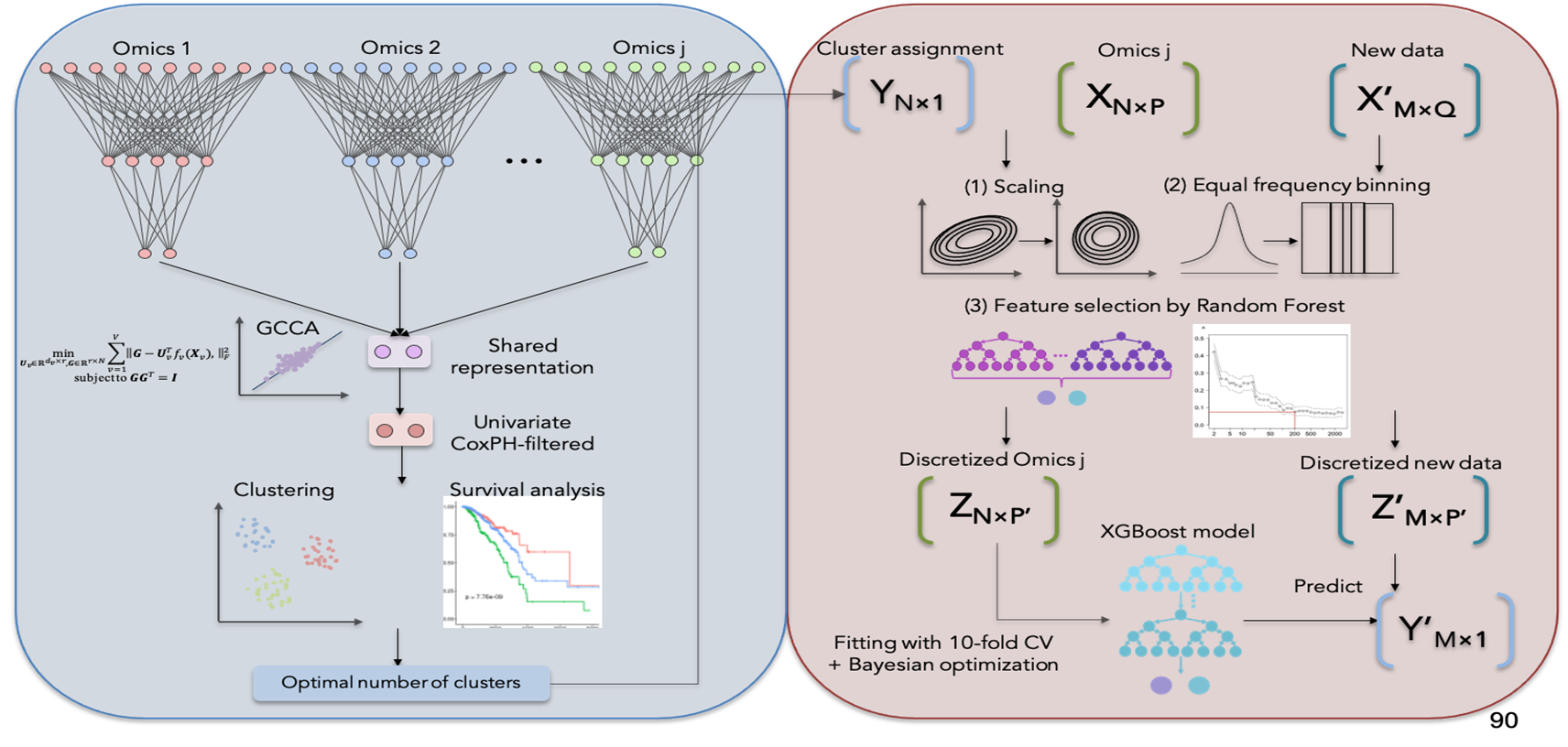 DeepMOIS-MC (Deep Multi-Omics Integrative Subtyping by Maximizing Correlation), a novel deep learning-based method that achieves multi-omics integration and subtyping of cancer by finding a low-dimensional shared representation that maximizes the correlation between multiple views. DeepMOIS-MC extends DGCCA (Deep Generalized Canonical Correlation Analysis), a canonical correlation analysis-based algorithm that can simultaneously learn nonlinear relationships between more than two views. The hypothesis is that the shared embedded space that maximizes correlation between views should contain most useful information for robust subtyping, since this indicates that certain patterns are repeatedly seen across multiple -omics platforms. We show that DeepMOIS-MC is indeed capable of robustly and accurately identifying cancer subtypes with enhanced prognostic stratification that are translatable across platforms.
DeepMOIS-MC (Deep Multi-Omics Integrative Subtyping by Maximizing Correlation), a novel deep learning-based method that achieves multi-omics integration and subtyping of cancer by finding a low-dimensional shared representation that maximizes the correlation between multiple views. DeepMOIS-MC extends DGCCA (Deep Generalized Canonical Correlation Analysis), a canonical correlation analysis-based algorithm that can simultaneously learn nonlinear relationships between more than two views. The hypothesis is that the shared embedded space that maximizes correlation between views should contain most useful information for robust subtyping, since this indicates that certain patterns are repeatedly seen across multiple -omics platforms. We show that DeepMOIS-MC is indeed capable of robustly and accurately identifying cancer subtypes with enhanced prognostic stratification that are translatable across platforms.
DNABERT: A BERT-based model for DNA-language in Human Genomes
Code
Yanrong Ji, Zhihan Zhou
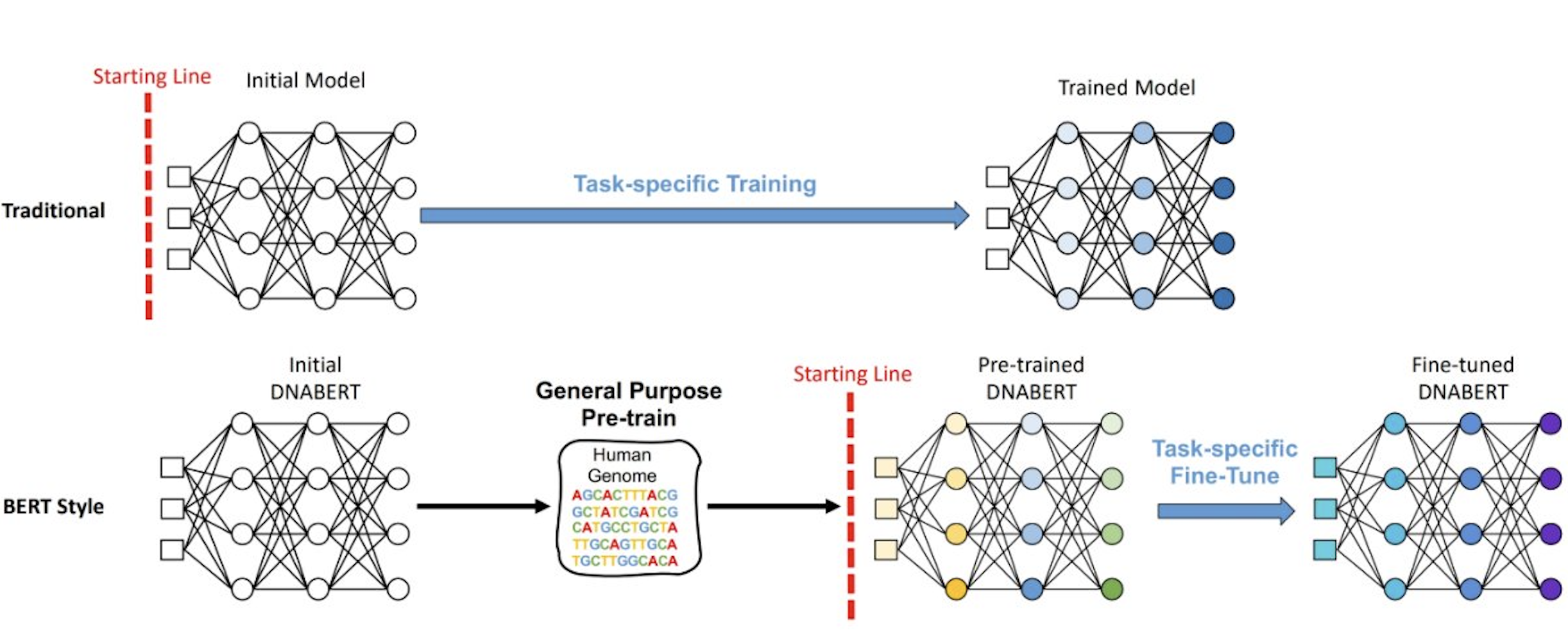 Understanding the hidden instructions within genome on gene regulation is crucial for biological research. However, complex language patterns widely exist in DNA, such as polysemy and distant semantic relationship, which previous methods often fail to capture especially in data-scarce scenarios. For the first time, our group (in collaboration with Dr Han Liu, Department of Computer Science, Northwestern University) developed DNABERT to enhance the global understanding of genomic sequences based on up and downstream sequence contexts. Using an innovative global contextual embedding of input sequences, DNABERT attempts to tackle the problem of sequence specificity prediction with a “top-down” approach by developing general understanding of DNA language via self-supervised pre-training and applying it to specific tasks (for example, prediction of promoters, transcription factor binding sites and splice sites), in contrast to the traditional “bottom-up” approach using task-specific data. Various modules of DNABERT are currently under development. It is anticipated that the pre-trained DNABERT on the human genome can also be readily applied to data from other organisms with exceptional performance.
Understanding the hidden instructions within genome on gene regulation is crucial for biological research. However, complex language patterns widely exist in DNA, such as polysemy and distant semantic relationship, which previous methods often fail to capture especially in data-scarce scenarios. For the first time, our group (in collaboration with Dr Han Liu, Department of Computer Science, Northwestern University) developed DNABERT to enhance the global understanding of genomic sequences based on up and downstream sequence contexts. Using an innovative global contextual embedding of input sequences, DNABERT attempts to tackle the problem of sequence specificity prediction with a “top-down” approach by developing general understanding of DNA language via self-supervised pre-training and applying it to specific tasks (for example, prediction of promoters, transcription factor binding sites and splice sites), in contrast to the traditional “bottom-up” approach using task-specific data. Various modules of DNABERT are currently under development. It is anticipated that the pre-trained DNABERT on the human genome can also be readily applied to data from other organisms with exceptional performance.
ExTraMapper: Exon- and transcript-level Mappings for Orthologous Gene Pairs
Code
Abhijit Chakraborthy, Ferhat Ay
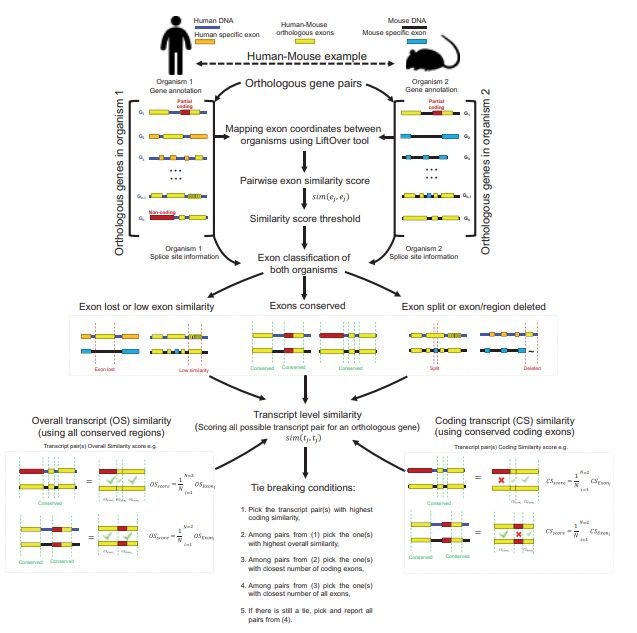 Access to large-scale genomics and transcriptomics data from various tissues and cell lines allowed the discovery of wide-spread alternative splicing events and alternative promoter usage in mammalians. Between human and mouse, gene-level orthology is currently present for nearly 16k protein-coding genes spanning a diverse repertoire of over 200k total transcript isoforms. We developed a novel tool, ExTraMapper, Exon and Transcript-level Mappings of a given pair of orthologous genes between two organisms leveraging sequence conservation between exons of a pair of organisms and produces a fine-scale orthology mapping at the exon and then transcript level. ExTraMapper identifies more than 350k exon mappings, as well as 30k transcript mappings between human and mouse using only sequence and gene annotation information. The tool identifies a larger number of exon and transcript mappings compared to previous methods. Further, it identifies exon fusions, splits and losses due to splice site mutations, and finds mappings between microexons that were previously missed. By reanalysis of RNA-seq data from 13 matched human and mouse tissues, we show that ExTraMapper improves the correlation of transcript-specific expression levels suggesting a more accurate mapping of human and mouse transcripts.
Access to large-scale genomics and transcriptomics data from various tissues and cell lines allowed the discovery of wide-spread alternative splicing events and alternative promoter usage in mammalians. Between human and mouse, gene-level orthology is currently present for nearly 16k protein-coding genes spanning a diverse repertoire of over 200k total transcript isoforms. We developed a novel tool, ExTraMapper, Exon and Transcript-level Mappings of a given pair of orthologous genes between two organisms leveraging sequence conservation between exons of a pair of organisms and produces a fine-scale orthology mapping at the exon and then transcript level. ExTraMapper identifies more than 350k exon mappings, as well as 30k transcript mappings between human and mouse using only sequence and gene annotation information. The tool identifies a larger number of exon and transcript mappings compared to previous methods. Further, it identifies exon fusions, splits and losses due to splice site mutations, and finds mappings between microexons that were previously missed. By reanalysis of RNA-seq data from 13 matched human and mouse tissues, we show that ExTraMapper improves the correlation of transcript-specific expression levels suggesting a more accurate mapping of human and mouse transcripts.
Platform-independent Isoform-level Gene Signatures for Stratification of Cancer Patients into Molecular Subgroups
Sharmishtha Pal, Yingtao Bi, Arunima Shilpi, Yanrong Ji, Manoj Kandpal
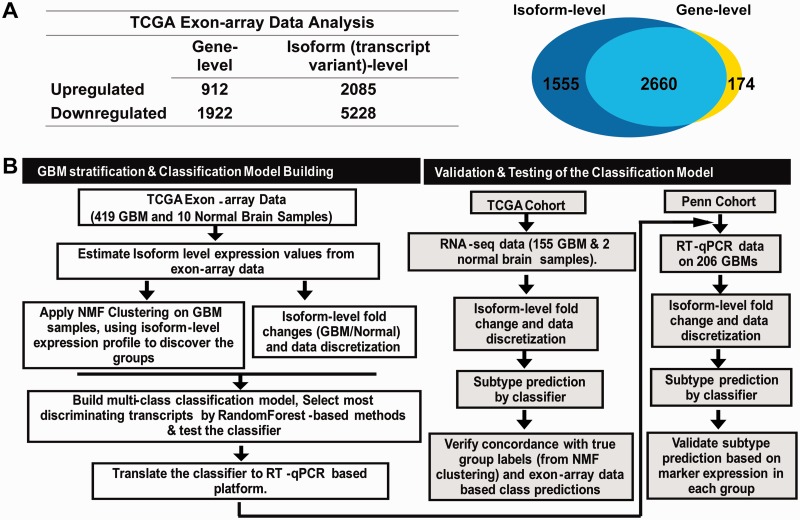 Based on recent studies from Our group and others, significant expression differences were observed between different sample groups (e.g., developmental stages, cancer subtypes, normal vs cancer) for numerous genes at the isoform-level but not at the overall gene-level. We investigated whether the isoform-level transcriptome changes could provide better patient stratification in terms of overall prognosis and classification accuracy. His group developed novel methods, by integrating data discretization, feature selection, and meta-classification algorithms, for derivation of platform-independent gene signature for multi-label molecular stratification of cancer patients, from exon-array and RNA-seq data. The application of these algorithms has led to the development of new methods for diagnosis of glioblastoma and ovarian serous carcinoma among others, and the investigation of alternative splicing on drug-target gene interactions.
Based on recent studies from Our group and others, significant expression differences were observed between different sample groups (e.g., developmental stages, cancer subtypes, normal vs cancer) for numerous genes at the isoform-level but not at the overall gene-level. We investigated whether the isoform-level transcriptome changes could provide better patient stratification in terms of overall prognosis and classification accuracy. His group developed novel methods, by integrating data discretization, feature selection, and meta-classification algorithms, for derivation of platform-independent gene signature for multi-label molecular stratification of cancer patients, from exon-array and RNA-seq data. The application of these algorithms has led to the development of new methods for diagnosis of glioblastoma and ovarian serous carcinoma among others, and the investigation of alternative splicing on drug-target gene interactions.
Isoform-level Gene Expression and Regulation in Mammalian Development and Cancer
Recent genome-wide studies have discovered that majority of human genes produce multiple transcript-variants/protein-isoforms, which could be involved in different functional pathways. Moreover, altered expression of specific isoforms for numerous genes is linked with cancer and its prognosis, as cancer cells manipulate regulatory mechanisms to express specific isoforms that confer drug resistance and survival advantages. For example, cancer-associated alterations in alternative exons and splicing machinery have been identified in cancer samples, suggesting that specific transcript-variants could be more effective as diagnostic and prognostic markers than corresponding genes. In a recent study, Our Dgroup discovered that majority of genes associated with neurological diseases expressed multiple transcripts through alternative promoters by using integrative NextGen sequencing based experimental approaches and bioinformatics analysis. The study also observed aberrant use of alternative promoters and splice variants in different cancers. Subsequently, his group demonstrated that cancer cell-lines regardless of their tissue of origin can be effectively discriminated from non-cancer cell-lines at isoform-level, but not at gene-level. The novel informatics methods have been successfully applied by his collaborators in different cancer studies.
Algorithms and Bioinformatics Software for Analyses of NextGen Sequence Data
Mapping genome-wide data to human subtelomeres has been problematic due to the incomplete assembly and challenges of lowcopy repetitive DNA elements. Our group developed novel bioinformatics pipelines for incorporating multi-read mapping for annotation of the updated assemblies using short-read data sets from ChIP-seq data, and RNA-seq data. As part of other collaborative efforts, we also developed bioinformatics methods for identification of single-nucleotide polymorphisms (SNPs) that alter miRNA gene regulation and influence tumor susceptibility.